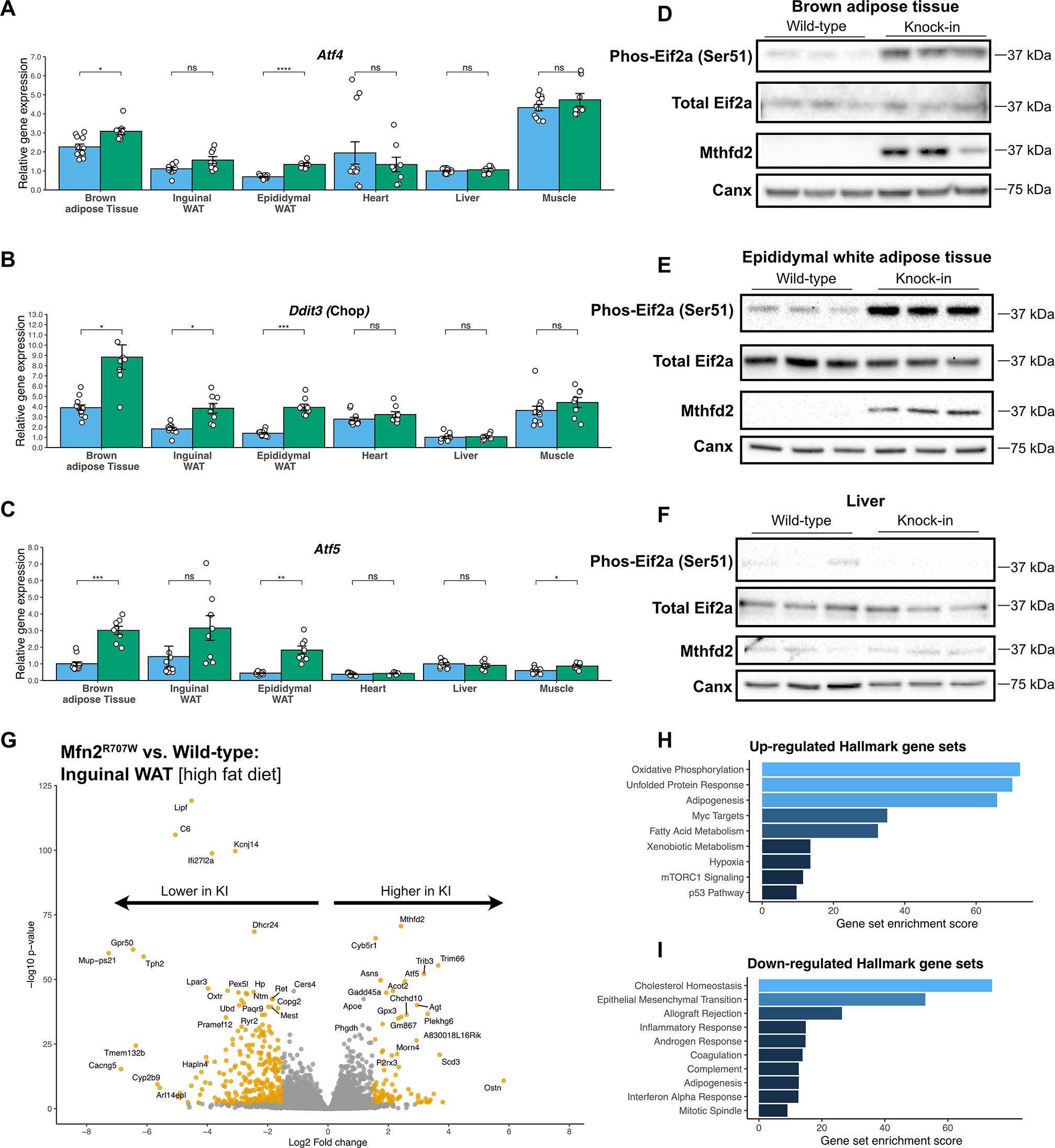
Learning how to do a GWAS meta-analysis
I feel proud that this month we’re publishing a large-scale GWAS meta-analysis identifying new candidate genes for hepatocellular carcinoma (HCC, or ‘liver cancer’) in adults.
You may reasonably be asking why I’m doing a project on adult liver cancer when I’m a paediatric hepatologist. I don’t look after adults, and cancer isn’t really my primary speciality either. The honest answer is that an enthusiastic medical student (now doctor) got in touch, keen to take on a project, and I thought this one was feasible and interesting. That was more than two years ago and, as always, it turned out to be harder than I’d anticipated.
This is one of the largest GWAS meta-analyses of HCC to date, and through a single-stage approach we’ve identified several novel genetic regions (loci).

This figure is a ‘Miami plot’, which shows chromosomes on the x-axis and each dot is a different genetic variant. The further up (or down i.e. away from the centre line) the more significantly the genetic variant is associated with liver cancer. The very significant variants stack up in lines, which I’ve then coloured in red (significant) or orange (suggestive). The top half uses one statistical model (MR-MEGA) and the bottom uses a different model (METAL).
A few things to points out:
Most of the controls don’t have liver disease. Almost all the controls in this meta-analysis are population-level controls who, on the whole, don’t have liver disease. But liver cancer most often develops in the context of chronic liver disease, particularly cirrhosis. So our HCC GWAS also shows genes related to cirrhosis rather than to cancer per se. Many of those signals sit in genes implicated in lipid droplets, which likely reflects that at a population level (particularly across Europe and North America) the main drivers of liver cancer relate to fat deposition in the liver, both metabolic (MASLD) and alcohol-related.
Some new signals look independent of cirrhosis. More interestingly, we found signals that appear independent of any effect on cirrhosis. One sits on chromosome 8, in what is actually a “gene desert”: a stretch with no genes in it. This region has been linked to several other cancers, and working out exactly which gene is responsible is difficult. Previous researchers studying other conditions, such as colon cancer, have shown that the DNA here can form loops, so that variants in this region physically interact with a distant stretch of DNA controlling levels of a cancer-related gene called MYC. We haven’t directly proven a role for MYC in liver cancer in this paper, but we’ve highlighted a region that has previously been tied to it so that’s where our suspicions lie.
Evidence for the beta-catenin pathway. MYC is also interesting partly because it sits downstream of the Wnt/β-catenin pathway, which is well-established in pro-cancer signalling. We found several other genes contributing to this pathway, both at genome-wide significance and just below it. This isn’t entirely novel biology as somatic mutations in this pathway are found in almost all forms of liver cancer but I think it’s quite elegant to see them all linked together in a single GWAS.
Some genes affect certain ancestries more than others. We also run an ancestral heterogeneity analysis, which let us show that some genetic variants have opposing effects across cohorts of different genetic ancestries. One clear observation was the lack of data in individuals of certain ancestries, including those of African or Caribbean descent. Again, this isn’t a novel obeservation in genetic epidemiology, and there are many good efforts trying to address it. I hope subsequent meta-analyses can include greater numbers of people from these ancestries, both for a more accurate picture of the genetics and for more equitable research across populations.
Finally, this project was a major source of personal development. I repeatedly feel as though I know how to code, then attempt a substantially more complicated paper and realise how little I actually knew. Then the cycle repeats with the next, harder project. This one involved a huge learning curve: using summary statistics, deriving our own novel GWAS summary stats from the All of Us dataset, an enormous amount of quality control, and a range of post-GWAS analyses. It’s reassuring to know I now have those skills, can run robust analyses, and will get to apply them to different questions in future.
PS — on the journal. This might look like a slightly obscure place to publish the paper, and I think that’s for a few reasons. First, there’s a preprint of another very large meta-analysis of cirrhosis and liver cancer, which I imagine will land in a very good journal with a short name. Second, several of the primary liver journals seemed unhappy with our lack of a separate validation cohort. I argued that our whole methodology was designed to maximise power in a single-stage meta-analysis but we couldn’t get into a higher-impact pure-genetics journal. So that’s how it ended up here. Either way, I’m proud of the work. I think it’s accurate, I think it’s true, and I hope it contributes to the literature in a positive way.